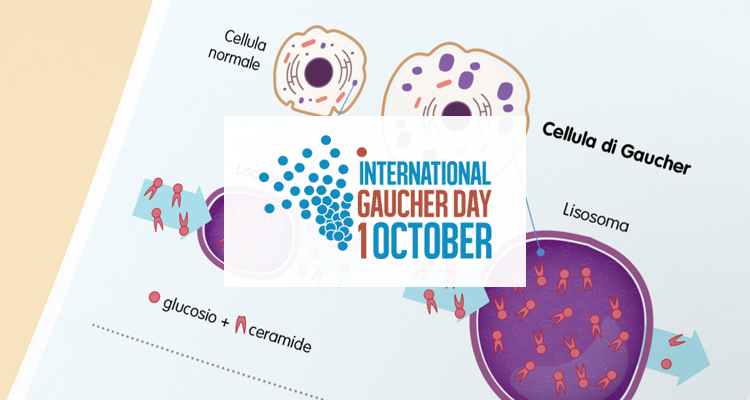
In Europa, una malattia che colpisce meno di una persona ogni 2.000 è considerata rara. Esistono tra le 6 mila e le 8 mila malattie rare nel mondo. Tra queste c’è la malattia di Gaucher, una patologia metabolica causata dalla mancanza di un enzima che permette alle cellule di digerire i prodotti di scarto del metabolismo. Gli scarti si accumulano così all’interno delle cellule e provocano problemi di salute in diverse parti dell’organismo.
___
In occasione della Giornata internazionale della malattia di Gaucher – celebrata ogni anno il primo ottobre – GIMEMA informazione ha intervistato Fiorina Giona, ematologa al dipartimento di Medicina traslazionale e di precisione all’università La Sapienza di Roma: “Sebbene sia genetica ed ereditaria, la malattia di Gaucher può manifestarsi a qualunque età con sintomi molto variabili, a seconda degli organi colpiti. Si distinguono 3 sottotipi: il tipo 1, più frequente, può comparire in età adulta e ha sintomi simili a quelli di una malattia ematologica; e i tipi 2 e 3 che manifestano invece sintomi neurologici con andamento diverso”.

Fiorina Giona, ematologa al dipartimento di Medicina traslazionale e di precisione all’università La Sapienza di Roma
Quante persone colpisce la malattia di Gaucher nel mondo? Quante in Italia?
Una malattia può essere rara in una regione, e frequente in un’altra. Il tipo 1, nel mondo, colpisce una persona su 40-60 mila, ma per esempio è molto frequente negli ebrei ashkenaziti tra i quali colpisce uno su 800 nati. La forma di tipo 2 è rarissima (1 su 100.000 nati) e la forma di tipo 3 colpisce una su 50-100 mila persone. È difficile conoscere il numero di persone affette in Italia. È stato calcolato che la malattia colpisce una persona su 40-86 mila persone, ma nelle regioni dove si fa lo screening neonatale l’incidenza è maggiore (fino a un caso su 20 mila).
Perché può essere considerata una malattia del sangue?
Perché le cellule in cui si accumula il materiale (i macrofagi) sono cellule del sangue. Anche per questo il tipo 1 spesso viene confusa con una malattia ematologica. I sintomi sono vari: addome persistentemente gonfio, difficoltà digestive e senso di pienezza, dovuti a un progressivo aumento di volume della milza, accompagnato a volte da quello del fegato; comparsa di dolori ossei o di fratture spontanee nell’adulto; alterazioni scheletriche e ritardo nella crescita nei bambini. Altri sintomi sono: stanchezza dovuta ad anemia e tendenza a sanguinare, dovuto alla riduzione del numero delle piastrine.
Quali sono oggi le terapie utilizzate per trattarla?
La prima terapia disponibile prevedeva la somministrazione per endovena dell’enzima mancante, ricavato dalla placenta umana che ne è molto ricca. Oggi sono disponibili tre tipi di enzimi prodotti in laboratorio con tecniche di biologia molecolare. In USA e Israele sono autorizzati tutti e 3, mentre in Europa e in Italia solo 2. Si tratta quindi di una terapia che può essere fornita solo dall’ospedale. Più recente è un altro tipo di trattamento, che impedisce la formazione della sostanza che l’enzima mancante dovrebbe digerire. Il vantaggio è che questa terapia – chiamata terapia di riduzione del substrato – può essere assunta per via orale, ma possono riceverla solo i pazienti che non eliminano il farmaco troppo velocemente. In Italia sono molti i centri specializzati particolarmente preparati nel trattamento della malattia di Gaucher, distribuiti uniformemente su tutto il territorio italiano. Per esempio il reparto di Pediatria all’ospedale Regina Margherita di Torino, l’ospedale Gaslini di Genova, il Policlinico di Milano e il Santa Maria della Misericordia di Udine nel Nord Italia. Il Bambin Gesù, il Policlinico a Roma, e i centri di Napoli e Catania per il Centro Sud.
Quali sono le nuove frontiere per la cura di questa malattia?
Purtroppo, tutte le terapie oggi disponibili sono efficaci solo per la malattia di Gaucher di tipo 1. Ma non in tutti i pazienti la terapia ha gli effetti sperati. Per questo negli ultimi anni la ricerca si è concentrata sulla terapia genica e su alcune piccole molecole, chiamate chaperones, che agiscono sugli enzimi anomali, facendoli funzionare. Queste molecole potrebbero funzionare anche nelle forme di malattia di Gaucher di tipo 2 e 3.
Che affetti ha avuto l’epidemia COVID-19 sui pazienti affetti da malattia di Gaucher?
La pandemia potrebbe avere un impatto negativo sull’accesso alle terapie. Per questo un gruppo di ricercatori italiani ha condotto uno studio su 250 pazienti per capire se la pandemia abbia condizionato il trattamento e l’accesso ai centri specialistici. Solo 11 pazienti hanno ridotto il numero di infusioni limitando l’accesso all’ospedale. La maggior parte dei pazienti ha continuato la terapia endovenosa in ospedale e, dove possibile, a domicilio; oppure ha continuato a prendere o ha iniziato il trattamento con il farmaco orale. Per quanto riguarda l’infezione da COVID-19, solo un paziente ha riportato sintomi lievi ma si è ripreso senza conseguenze. Lo studio verrà presentato al prossimo congresso WORLD Symposium, a febbraio 2021.
Il tema di quest’anno della Giornata internazionale è proprio la terapia domiciliare. Qual è la situazione in Italia?
Purtroppo, la terapia domiciliare non è possibile né è garantita in tutte le regioni italiane. Il servizio è attivo in Lazio, Veneto, Campania, Puglia, Sardegna, Abruzzo, Lombardia, Liguria, Sicilia, Calabria e Umbria. In Toscana e Marche è stato attivato con l’emergenza COVID-19 mentre in Piemonte viene attivato solo per casi particolari. Quattro regioni – Emilia Romagna, Molise, Trentino Alto Adige e Valle d’Aosta – non hanno alcun tipo di servizio domiciliare. Le aziende farmaceutiche produttrici della terapia enzimatica offrono gratuitamente il servizio infermieristico domiciliare, che viene utilizzato dalla maggior parte delle regioni. L’ideale sarebbe che il Servizio Sanitario garantisse la terapia domiciliare su tutto il territorio nazionale, in modo che tutti i malati con Gaucher possano ricevere lo stesso trattamento ovunque si trovino.